

Biomacromolecules such as oligonucleotides and peptides are sought after for construction of novel nanostructures for the solution of a variety of problems in medicine. The use of oligonucleotides to prepare well-defined nanostructures utilizing Watson–Crick base paring was proposed over two decades ago.1 Since then, the concept has evolved into an exciting field demonstrating the preparation of several novel two-dimensional and three-dimensional structures in a wide range of applications, including delivery, sensing, imaging, assembly, and immunostimulation.2–8In vitro and in vivo drug and gene delivery studies using DNA origami were also successfully demonstrated.9–15
Transfection methods for gene regulation were widely investigated after the discovery of RNAi.16 The DNA nanotechnology concept has shown promising results for the transfection of siRNA and antisense oligonucleotides by embedding them into DNA-based structures. For example, phosphorothioate antisense oligonucleotides were incorporated into triangular prism-shaped DNA cages by stapling oligonucleotides complementary to the antisense oligonucleotides through hybridization.13 This simple strategy allowed incorporation of 1, 2, 4, or 6 antisense oligonucleotides into the DNA cages. Then, the whole structure was delivered into Hela cells using liposomes for silencing of luciferase expression.
In another study, one-dimensional periodic DNA structures called nanoribbons were constructed, and their surface was modified with siRNA for targeting survivin mRNA in human ovarian cancer cells.14 The siRNA conjugated nanoribbons successfully silenced the survivin expression. In vivo gene silencing using DNA nanostructures was also reported.15 Prism-shaped tetrahedral DNA nanostructures modified with three folate molecules were used for in vivo siRNA delivery. Longer blood circulation time and efficient knockdown of GFP and luciferase genes were achieved by using externally labeled DNA nanoparticles with siRNA and folate.
To increase target specificity and affinity in gene regulation studies, oligonucleotide backbone is modified with certain chemical groups to generate synthetic antisense oligonucleotides. These oligonucleotides include morpholinos, methylphosphonates, phosphorothioate nucleic acids (NAs), peptide NAs, and a number of others.17
Morpholino, an antisense oligonucleotide, synthesized by linking morpholine rings through phosphorodiamidate groups instead of phosphates, is preferred in silencing studies due to their excellent biological stability, which overcomes the restraint of substantial degradation of bare nucleic acids in biological systems, and high sequence specificity.18 Morpholino antisense oligonucleotides also have other superior features than their counterparts, including high solubility in aqueous media, and chemical stability.19 In addition, their uncharged backbone prevents their nonspecific interactions with cellular proteins giving them the advantage of being free from off-target effects.20
Compared with the other nonviral delivery systems, cationic polymers21 and cationic lipids22 are more widely used for siRNA delivery. The cationic delivery vehicles naturally interact through electrostatic interactions between negatively charged phosphate backbone of NA and positively charged groups of carrier.23,24 Since morpholino antisense oligonucleotides do not carry charge, their packing with positively charged carriers may not be efficient,25 which limits in vitro application of morpholinos. Delivery of morpholinos into cultured cells was studied with scrape loading26 and osmotic delivery.27 However, both methods have the limitations of not being available for many cell types and effectiveness.28
DNA nanotechnology approach allows incorporation of any types of artificial oligonucleotides into DNA-based nanostructure through complementary base pairing. Antisense oligos can be perfectly integrated into DNA nanostructures due to their similar chemistry.29 Unlike other antisense oligos, morpholino/DNA hybridization occurs efficiently independent from salt concentration,19 making DNA nanostructures perfect candidates for delivery of morpholinos in vitro and in vivo.
Since the cellular uptake of negatively charged species is highly restricted, DNA-tiles are not effectively uptaken by cells. Thus, we incorporated the gold nanoparticles (AuNPs) with an average size of 13 nm into the DNA-tile structure since oligonucleotide-coated AuNPs are effectively uptaken by cells.30 The AuNPs coated with oligonucleotides complementary to the sticky ends of the tile were hybridized to the three sticky ends of the tile. Note that the safety and biocompatibility of oligonucleotide-coated AuNPs were studied, and found that they caused no toxic cellular responses.31
In this study, the morpholinos were specifically designed to regulate human breast cancer genes HER2 and ERα. In the proof-of-concept experiments, each morpholino antisense oligonucleotide was embedded into a separate DNA-tile-AuNPs carrier, and expressions of the mammalian genes were successfully inhibited in human cancer cells. First, the effective DNA-tile concentration was determined. Then, the transfection of the cells with morpholino embedded DNA-tile-AuNPs was studied by evaluating the expression of targeted genes. The effects of gene silencing on cellular proliferation and cell cycle were also investigated. The efficacy of DNA-tile-AuNPs was compared with a cationic lipid-based commercial carrier, and the results showed that DNA-tile-AuNPs downregulated the genes more effectively. The nanostructure was able to transfer at higher antisense concentrations at which the liposome could be toxic.
Materials and Methods
Reagents
BT-474 and MCF7 breast cancer cell lines were purchased from the American Type Culture Collection (Manassas, VA), MCF-10A cells were kindly provided by Fikrettin Şahin Lab. DreamFect Gold (OzBiosciences, Marseille, France). All oligonucleotides were obtained from Alpha DNA (Montreal, Canada). The morpholino antisense oligonucleotides against HER2 and ERα genes were designed and synthesized by Gene Tools.
Preparation of DNA-tile

Two different morpholinos were embedded into DNA-tiles separately (see Table 1 for primer numbers used for construction of DNA-tiles targeting HER2 or ERα genes). DNA-tile structures were prepared as explained in our previous study.12 In brief, DNA-tile structures were constructed using one scaffold oligonucleotide and eight complementary helper oligonucleotides, including the morpholino sequences, which are provided in Table 2. Equimolar amounts of the oligonucleotides were added in TAE/Mg2+ hybridization buffer, which includes 40 mM Tris, 20 mM acetic acid, 2 mM ethylene diamine tetra acetic acid (EDTA), and 12.5 mM magnesium acetate pH 8.0. This mixture was heated to 95°C, and then slowly cooled to room temperature once. No further heating and cooling procedures were applied.
 DNA-tile nanostructures were prepared upon bottom-up strategy by using self-organization properties of nucleic acid molecules. The oligonucleotides were denatured at 95°C, and cooled very slowly. During the temperature decrease, the complementary oligonucleotides connected to the scaffold DNA at their annealing temperature and folded into a tile-shaped DNA nanostructure. First, the four 47 base long oligonucleotides were annealed, and then the 25 base long oligonucleotides, including morpholino, were incorporated to the structure by complementary base pairing resulting DNA-tile with a theoretical size of 28 nm and with 8 sticky ends. These sticky ends were further used for AuNPs modifications.
DNA-tile nanostructures were prepared upon bottom-up strategy by using self-organization properties of nucleic acid molecules. The oligonucleotides were denatured at 95°C, and cooled very slowly. During the temperature decrease, the complementary oligonucleotides connected to the scaffold DNA at their annealing temperature and folded into a tile-shaped DNA nanostructure. First, the four 47 base long oligonucleotides were annealed, and then the 25 base long oligonucleotides, including morpholino, were incorporated to the structure by complementary base pairing resulting DNA-tile with a theoretical size of 28 nm and with 8 sticky ends. These sticky ends were further used for AuNPs modifications.
Agarose gel electrophoresis
Agarose gel electrophoresis was performed to monitor the formation and the serum stability of DNA-tile structures. For monitoring the formation of the nanostructure, nine samples were prepared. The first sample contained 1 μM of one oligonucleotide (P1) and for the other samples 1 μM of each complementary oligonucleotide was added sequentially into TAE/Mg2+ hybridization buffer up to all nine complementary oligonucleotides. For determination of serum stability, DNA-tile nanostructures were incubated in serum containing cell medium for 6, 24, 36, and 48 h. 0.2 μM of the incubated nanostructures was loaded to the gel. A 2% agarose gel was prepared in 1 × TAE buffer, and ethidium bromide was added into gel for visualization of DNA samples. The samples were run at 90V for 30 min. The agarose gel images were taken under ultraviolet (UV) light by a Bio-Rad gel imaging system.
Synthesis of AuNPs
The AuNPs with an average size of 13 nm were synthesized with Turkevich method.32 In brief, 40 mg of gold chloride (HAuCl4.3H2O) was dissolved in 100 mL ddH2O. The prepared solution was heated and stirred until boiling. While heating, 10 mL of 38.8 mM sodium citrate (Na3C6H5O7) solution, which induces reduction of gold salt, was quickly added into the boiling solution. It was kept boiling for 15 min, and left to cool at room temperature.
Coating AuNPs with oligonucleotides
Oligonucleotide modification of AuNPs (Oligo-AuNPs) was carried out according to the salt aging method with a few modifications.33 In brief, 1 mL of 13 nm AuNPs (400 μg/mL) was mixed with 0.1 M potassium phosphate buffer at pH 7.4 and 0.125 mL of 0.1% sodium dodecyl sulfate (SDS) by bringing the final concentrations at 0.01 M phosphate buffer and 0.01% SDS.
A 10 μL of 100 μM 5′-thiol modified 14-mer poly-thymine oligonucleotide (5′-SH-TTTTTTTTTTTTTT) was added to the mixture, and the final mixture was gently mixed for 30 min at room temperature. 0.1 mL of NaCl solution was added from 2 M NaCl stock solution at three steps and the final NaCl concentration was brought to 0.15 M. At the every step of NaCl addition, the suspension was sonicated for 20 s and mixed for 30 min. After the third addition of NaCl solution, the suspension containing AuNPs was left shaking overnight. The process was followed by centrifugation of the suspension at 13,000 rpm for 20 min to remove the excess oligonucleotides. The supernatant was replaced with ddH2O. The washing step was repeated three times. The pellet was dissolved in ddH2O, and the suspension was stored at 4°C.
Construction of DNA-tile-AuNPs nanostructure
The DNA-tile-AuNPs nanostructures were constructed by mixing ∼5.4 × 1013 Oligo-AuNPs with 1.8 × 1013 DNA-tiles containing suspension in TAE/Mg2+ hybridization buffer. The mixture was incubated overnight at room temperature. The schematic presentation of DNA-tile modification with Oligo-AuNPs is shown in Figure 1.

UV/vis spectroscopy
The absorbance spectra of the 1 nM suspensions containing 13 nm AuNPs, Oligo-AuNPs, and DNA-tile-AuNPs were registered using a UV/vis spectrometer (PerkinElmer).
Dynamic light scattering
The hydrodynamic sizes of 13 nm AuNPs, Oligo-AuNPs, and DNA-tile-AuNPs were analyzed at 25°C using Nanozetasizer (Malvern Instruments).
Affinity of AuNPs to DNA-tiles
The affinity of Oligo-AuNPs for DNA-tiles was tested using agarose gel electrophoresis. DNA-tile-AuNPs structures were incubated in fetal bovine serum (FBS) containing culture medium for 24 and 48 h. Two percent agarose gel was prepared in 1 × TAE buffer (pH 8.2). Bare AuNPs, Oligo-AuNPs, DNA-tile-AuNPs without incubation, DNA-tile-AuNPs incubated for 24 and 48 h were centrifuged at 13,000 rpm for 20 min, the supernatant was removed and 15 μL of remaining pellet was loaded on agarose gel. The samples were run at 80V for 1 h. The image of the gel was taken using a digital camera.
Cell lines and culture
BT-474 and MCF7 breast cancer cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) high glucose (10% FBS, 1% penicillin-streptomycin) cell medium, MCF-10A cells were cultured with MEGMTM Mammary Epithelial Cell Growth Medium at 37°C under 5% CO2 atmosphere.
Cellular uptake studies
Confocal microscopy
Internalization and localization of the gene silencing nanostructure were determined using confocal microscopy. The primer number P10 was ordered with a 5′-Cy5 modification. For internalization experiments, the dye-modified oligonucleotide was used for construction of the DNA-tile-AuNPs structure. Glass cover slips were sterilized with 70% ethanol and placed in six-well plates and 100,000 BT-474 cells were seeded in each well. Attached cells were treated with Cy5-labeled DNA-tile-AuNPs nanocarriers and incubated for 2 h. The cells were fixed using 2% paraformaldehyde solution for 20 min at +4°C and stained with lysotracker green DND-26 (Thermo Scientific) at room temperature. The samples were washed with phosphate-buffered saline (PBS) three times for 5 min and immediately analyzed using a confocal microscope (Carl Zeiss LSM 700, Oberkochen, Germany) to visualize endosomes/lysosomes (green fluorescence), and Cy5-DNA-tile-AuNPs (red fluorescence).
Flow cytometry
A total of 40,000 MCF7 cells were seeded in six-well plates and incubated for 24 h before incubation with Cy5-DNA-tile-AuNPs nanostructures. The nanocarrier system was added to the cells at 30 nM concentration and incubated in cell culture conditions for 2 and 4 h. The cells were washed with PBS three times, detached using trypsin-EDTA (0.25%), and collected by centrifugation at 2,000 rpm for 5 min. After the three more washing steps, cells were collected and dissolved in PBS. The harvested cells were immediately analyzed using Guava EasyCyte flow cytometer (MerkMilipore) to investigate red fluorescence intensity of the cells.
Inductively coupled plasma mass spectroscopy
Inductively coupled plasma mass spectroscopy (ICP-MS) technique was used to evaluate the internalization of DNA-tile-AuNPs by determination of the presence of intracellular Au ions. A total of 40,000 MCF7 cells were seeded into six-well plates and incubated for 24 h for attachment. The cells were treated with DNA-tile-AuNPs containing 85 μg Au in 2 mL medium and incubated for 2, 6, and 24 h. Each treatment was repeated three times. At the end of the exposure times, the cells were detached and lysed using 200 μL lysis buffer (Thermo Scientific). The lysed cells were digested by adding 0.5 mL of freshly prepared aqua regia (highly corrosive) and incubated for 15 min. Then each sample volume was completed to 10 mL with dH2O and analyzed for Au content. 197Au isotopes were monitored using ICP-MS (XSeries 2; Thermo Fisher Scientific, Bremen, Germany). Each sample was measured three times.
Western blotting
BT-474 and MCF7 cells were seeded in six-well plates at 150,000 cells per well. After the attachment at the end of 24 h, the cells were washed with PBS and treated with DNA-tile-AuNPs in FBS-free DMEM at the concentrations between 20 and 40 nM, and effective concentration was determined. To assess the silencing efficiency of DNA-tile-AuNPs, a routinely used liposome system, DreamFect Gold (OzBiosciences) was employed. The cells were treated with 30 nM DNA-tile-AuNPs in a serum-free medium. The morpholino containing liposome treatments were performed using the reagent according to the instructions of the manufacturer. In brief, 10 μL of DreamFect Gold was dissolved in 0.5 mL of FBS-free medium, and then combined with 0.5 mL of morpholino containing medium at a final morpholino concentration of 30 nM and the mixture was incubated for 20 min at room temperature. The cells were treated with morpholino embedded DNA-tile-AuNPs and liposome for 4 h. The serum-free medium was replaced with FBS-enriched DMEM and incubated for another 24 h. Cell lysates were prepared using RIPA buffer (Santa Cruz Biotechnology, Inc.). Protein concentration of each sample was determined using Bradford Assay according to the instructions reported in reference.34 A 40 μg of total protein was subjected to denaturing polyacrylamide gel. The proteins were transferred to polyvinylidene fluoride (PVDF) membranes and placed into blocking solution (3% bovine serum albumin [w/v] in TBS-T) at room temperature for 1 h. The membranes were incubated with rabbit anti-HER2 primary antibody (Cell Signaling Technology, Inc.), rabbit anti-ERα primary antibody (Merck Milipore), mouse anti-Cyclin A2 primary antibody (Cell Signaling Technology, Inc.), rabbit anti-Cyclin E2 primary antibody (Cell Signaling Technology, Inc.), and Cyclin B1 primary antibody (Cell Signaling Technology, Inc. and all 1:1,000 dilutions as indicated) overnight at 4°C. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression was used as control of the equal amounts of protein loading. Anti-mouse immunoglobulin G (IgG), horseradish peroxidase (HRP)-linked antibody (Cell Signaling Technology, Inc.) and anti-rabbit IgG, HRP-linked antibody (Cell Signaling Technology, Inc.) were used as secondary antibodies. The western blotting images were taken using a ChemiDoc MP imaging system (BioRad). The analyses were performed with ImageJ software, and the band intensities were normalized to GAPDH and quantified with respect to controls.
Cell proliferation assay
MCF-10A cells were seeded in 96-well plates at the density of 5,000 cells per well and allowed to attach for 24 h. The suspension of DNA-tile-AuNPs was added into FBS-free DMEM at the morpholino concentrations between 0.6 and 80 nM. After 4 h incubation, the media was removed, and FBS completed DMEM was added. Then, the cells were left for incubation for 24 h. The cell viabilities were determined using 10% WST-1 dye.
For the analysis of proliferation inhibition after silencing, BT-474 and MCF7 cells were seeded in 24-well plates at 20,000 cells per well. The cells were treated with DNA-tile-AuNPs, only morpholino, and DNA-tile without morpholino with three repeats of each. One group of cells was treated only FBS-free culture medium as control. After 4 h incubation, the FBS-free medium was replaced with a completed cell medium. Seventy-two hours later, the cells were detached from the surface and counted. The dead cells were distinguished using trypan blue staining.
Cell cycle analysis
BT-474 and MCF7 cells were seeded in six-well plates at 150,000 cells per well and incubated for 24 h for attachment. The cells were transfected with 30 nM of DNA-tile-AuNPs nanostructures and incubated MCF7 cells for 48 h, and BT-474 cells for 120 h post-transfection by considering the cell cycle durations of each cell line. The cells were collected after trypsinization and centrifuged at 1,500 rpm at room temperature. The pellet was washed with PBS and fixed in 70% cold ethanol overnight at 4°C. After the fixation process, the cells were collected with centrifugation and incubated with RNase A (300 μg/mL) at 37°C for 30 min. Then, the cells were stained with propidium iodide dye and analyzed using Guava EasyCyte flow cytometer (MerkMilipore).
Results
Preparation and characterization of DNA-tile nanostructures
For preparation of DNA origami structures, mostly a viral genome and hundreds of staple strands are needed. The complicated formation of the structures makes DNA origami expensive and impractical for potential large-scale applications. In this study, we constructed a tile-shaped DNA nanostructure by using only nine complementary oligonucleotides (see Table 1), including a morpholino sequence with one-step hybridization as schematically represented in Figure 1. This design allows incorporating up to four copies of antisense oligonucleotides into one DNA-tile structure from its four arms. It is also possible to embed morpholino into the tile body, but it may take a longer time for release due to the required longer time for degradation of the tile. Thus, we preferred to place the morpholino to the arms.
The formation of the DNA-tile structures was monitored using agarose gel electrophoresis. In Figure 2A, the agarose gel electrophoresis image of the sequentially hybridized oligonucleotides is shown. The wells labeled with the primer numbers contain the increasing number of complementary oligonucleotides to form the tile structure. The first well had only one oligonucleotide, and the others were the samples prepared by adding sequential complementary oligonucleotides. The last sample was complete DNA-tile structure. As seen, the molecular weight of the hybridized structures gradually increases with the addition of sequential complementary oligonucleotides, which indicates the formation of the tile structure.

The incorporation of morpholinos into the tile structure was also monitored with agarose gel electrophoresis. Figure 2B shows the gel image of the DNA-tile without morpholino that is labeled as Tile, and morpholino embedded DNA-tile that is labeled as M. Owing to neutral charge of morpholino structure, the free morpholinos do not move through the agarose matrix. In contrast, the morpholinos embedded into the tile structures run together with the DNA-tile but migrate slower due to the increased weight. The agarose gel electrophoresis study indicates that morpholinos are successfully embedded into the DNA-tile structures.
Stability analysis of DNA-tile nanostructures
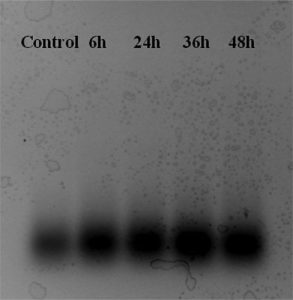
Stability of constructed DNA-tile structures was investigated. For this, the DNA-tiles were incubated in a cell medium supplemented with 10% FBS for 6, 24, 36, and 48 h. After the indicated time intervals, the samples were directly taken from the incubated environment and loaded onto agarose gel. Figure 3 shows the gel electrophoresis image of the tiles recovered from the cell culture medium. After incubation of DNA-tile nanostructures in serum containing medium, aggregations and protein corona caused higher band intensities than control sample. As seen, there is no indication that the tiles are disassembled in cell culture medium as indicated by intact spots on the gel electrophoresis image. One can easily conclude that the tiles are stable in cell culture medium and structural integrity of DNA-tile remained up to 48 h.
Preparation of DNA-tile-AuNPs
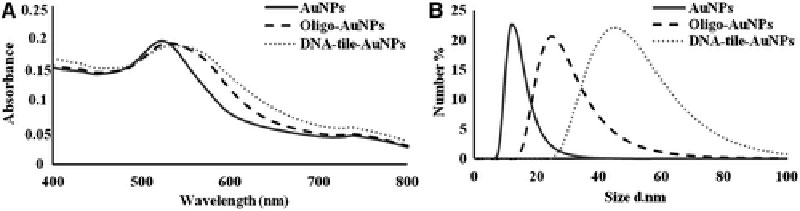
As it was mentioned earlier, DNA-tile structures were modified with 13 nm AuNPs to increase their intracellular uptake. The AuNPs were first coated with thiol modified oligonucleotides complementary to sticky ends of the tiles and then hybridized with the DNA-tile structure. The modification of DNA-tile structures with the AuNPs was characterized. UV/vis spectra of the suspension of AuNPs, Oligo-AuNPs and DNA-tile-AuNPs are shown in Figure 4A. The surface plasmon absorption band for 13 nm AuNPs colloidal suspension was at around 520 nm. With the oligonucleotide modification, the absorption maximum increased to 529 nm. When the absorbance spectrum of DNA-tile-AuNPs nanostructures was measured, it was at around 538 nm demonstrating the formation of targeted structure. The hydrodynamic sizes of the AuNPs, Oligo-AuNPs, and DNA-tile-AuNPs were determined using dynamic light scattering (DLS) and are shown in Figure 4B. The average hydrodynamic size of bare AuNPs was 13 nm. The average sizes of the Oligo-AuNPs and DNA-tile-AuNPs observed with DLS were about 24 and 51 nm, respectively. As seen, an increase in hydrodynamic radius was observed from bare AuNPs to DNA-tile-AuNPs supporting the results of UV/vis spectroscopy and indicating the formation of DNA-tile-AuNPs.
After preparation of DNA-tile-AuNPs nanocarriers, the affinity of AuNPs with the DNA-tile structures was investigated. DNA-tile-AuNPs carriers were incubated for 24 and 48 h in serum environment and bare AuNPs, Oligo-AuNPs and DNA-tile-AuNPs nanostructures before and after incubations were analyzed using agarose gel electrophoresis. Supplementary Figure S1 shows the gel image. Unmodified AuNPs aggregated immediately after loading to buffer containing well and are seen as black area into first well. The molecular weight difference between Oligo-AuNPs and DNA-tile-AuNPs indicated the effective binding of Oligo-AuNPs and DNA-tile-AuNPs. There was no indication of dissociation for the DNA-tile-AuNPs after incubations, which demonstrated high affinity of AuNPs for DNA-tiles.
Cellular uptake studies
Internalization of DNA-tile-AuNPs nanostructures was investigated using confocal microscopy, flow cytometry, and ICP-MS techniques. To determine the internalization way, the nanocarriers were labeled with Cy5 dye (Cy5-DNA-tile-AuNPs). One of the oligonucleotides (P10) was ordered with 5′-Cy5 modification, and the fluorescence labeled oligo was used for construction of DNA-tile-AuNPs. Breast cancer cells were treated with Cy5-DNA-tile-AuNPs nanostructures for 2 h and endosomes were stained using LysoTracker green. The breast cancer cells were analyzed with confocal microscopy. Supplementary Figure S2 shows the confocal microscopy images of BT-474 cells showing the cellular localization of endosomes/lysosomes (green) and Cy5-DNA-tile-AuNPs nanocarriers (red). The colocalization of green and red fluorescence in the merged image suggested the internalization of the nanocarrier system mediated by endosomes. Also nanocarriers localized in cytoplasm seen from the images indicated the escape from endosomal entrapment after 2 h incubation.
To follow the cellular internalization efficiency of the nanocarriers, MCF7 cells were treated with Cy5-DNA-tile-AuNPs nanostructures and incubated for 2 and 4 h. The cells were analyzed with flow cytometry at each time interval. The red fluorescence intensity of the cells was compared with untreated control cells, and the results are shown in Supplementary Figure S3A. The fluorescence intensity of the cells increased after incubation with the nanocarrier system, and fluorescence emission was detected at ∼95% of the treated cells after 4 h incubation demonstrating the efficient delivery of the nanocarrier system.
The internalization of DNA-tile-AuNPs was also followed by ICP-MS. A total of 40,000 MCF7 cells were treated with the nanostructure at Au dose of 42.5 μg/mL and incubated for 2, 6, and 24 h. After cellular uptake of DNA-tile-AuNPs carriers, the intracellular Au content was determined. Supplementary Figure S3B shows the ICP-MS results. The data showed that ∼60% of the Au content was internalized by cells after 24 h incubation.
Cell viability
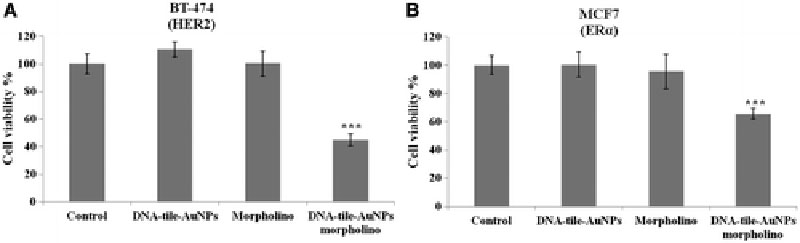
The cytotoxic effect of the morpholino against ERα embedded DNA-tile-AuNPs nanostructures was tested on a nontumorigenic mammary epithelial cell line MCF-10A at various doses between 0.6 and 80 nM. The viability of MCF-10A cells is shown in Figure 5. The results indicate that the DNA-tile-AuNPs has no toxic effect on the tested cells and it can be used for delivery of high concentrations of morpholinos without safety concerns.

Inhibition of gene expressions
The performance of DNA-tile-AuNPs was tested by monitoring the gene expression levels of targeting proteins. HER2 and ERα antisense morpholino embedded DNA-tile-AuNPs were tested on HER2 and ERα overexpressing BT-474 and MCF7 cells, respectively. Those cells were treated with increasing DNA-tile concentrations starting from 20 up to 40 nM DNA-tile-AuNPs. The minimum effective concentration for BT-474 cells was 30 nM, whereas the decrease in target gene expression in MCF7 cells was achieved even in 20 nM morpholino concentration. Figure 6A and B show that the silencing efficiency of DNA-tile-AuNPs depends on the dose, and it is observed that it decreased the expressed protein levels in the cells even at very low concentrations. The effective concentration for both cell lines was found as 30 nM, which was chosen as an optimal concentration for further experiments.

The efficiency of DNA-tile-AuNPs nanocarriers was compared against a conventional liposome-based system. For a comparative evaluation, the cells were treated with both DNA-tile-AuNPs and liposome at the selected morpholino concentrations. After a 24 h treatment, the protein expression levels were determined using western blotting analysis. As seen from the western blot images in Figures 6C and D, the DNA-tile-AuNPs decreased the expression of target genes more effectively than that of the liposome-based one.
Inhibition of breast cancer cell proliferation
Furthermore, the proliferation rates of BT-474 and MCF7 cells were determined according to the living cell numbers and compared with the untreated control groups after 72 h incubation. Figure 7 shows that HER2 antisense morpholino embedded DNA-tile-AuNPs led ∼50% inhibition of proliferation in BT-474 cells, whereas the proliferation rate in MCF7 cells decreased about 35% after treatment with ERα antisense morpholino embedded DNA-tile-AuNPs. These results also demonstrated the efficiency of DNA-tile-AuNPs-based approach on the silencing of genes overexpressed in breast cancers that induces uncontrolled cell proliferation.
Cell cycle analysis
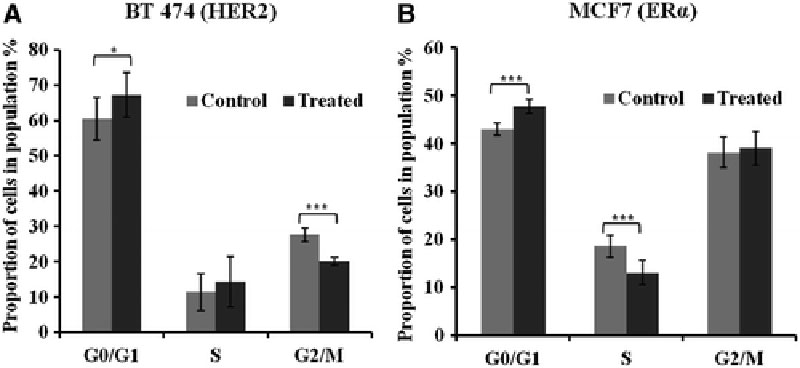
The influence of gene silencing by DNA-tile-AuNPs on the cell cycle function of breast cancer cells was determined using flow cytometry. BT-474 and MCF7 cells were treated with DNA-tile-AuNPs for the targeted silencing of HER2 and ERα, respectively, and their cell cycle phases were analyzed. Figure 8 shows the cell cycle results of untreated (control) cells and breast cancer cells treated with DNA-tile-AuNPs nanostructures against HER2 and ERα genes. Inhibition of HER2 and ERα expressions using the nanostructure caused a decrease in S and M phases at the cell cycle and an increase in the G0/G1 phase compared with controls. In BT-474 cells, the mean proportion of the cells in G0/G1phase increased from 60.5 to 67.2 (p < 0.05) and mean value of G2/M decrease from 27.7 to 20.1 (p < 0.001) after HER2 silencing. The percentage of MCF7 cells at G0/G1 increased from 43.1 to 47.8 (p < 0.001), and S phase decreased from 18.6 to 13.1 (p < 0.001) after inhibition of ERα gene. Expressions of cell cycle progression proteins cyclin E2, cyclin A2, and cyclin B1 were investigated after silencing of the HER2 and ERα genes and shown in Supplementary Figure S4. A decrease in cyclin E2 and cyclin A2 protein expressions was observed and cyclin B1 was downregulated robustly in BT-474 and MCF7 cells treated with DNA-tile-AuNPs.
The overall results showed that inhibition of expression of both proteins in targeted cells affected the cell cycle progression in G0/G1 phase and consistently inhibited the proliferation rate demonstrating the efficiency of DNA-tile-AuNPs nanocarriers for gene silencing.
Discussions
Our strategy was to develop a simple nonviral DNA-based gene delivery system by integrating the gene silencing oligos by complementary base pairing. In this structure, only nine oligonucleotides were practically assembled into a four-arm DNA-tile structure. From one of its sticky ends, it was stapled with the antisense morpholino, and the rest of the
arms were modified through hybridization of oligonucleotides attached on the AuNPs surface. The incorporation of the biocompatible AuNPs into the designed DNA-tile system can not only facilitate their intracellular uptake but also prevent their rapid enzymatic digestion.30,31 The intracellular uptake efficiency of the nanocarrier system was investigated, and effective internalization of the DNA-tile-AuNPs nanostructures was demonstrated.
Most of the delivery systems are susceptible to dissociation in the extracellular environment, so one of the major challenges for a gene delivery system is to retain structural integrity of the system and to protect the silencing oligonucleotides from external effects until the cellular internalization. Thus, we studied the stability of the DNA-tile-AuNPs structures in serum containing cell culture medium. The results showed that the DNA-tile-AuNPs nanostructures are resistant to serum degradation, and this will be an advantage for in vivo therapeutic applications.
To investigate the gene silencing performance of DNA-tile-AuNPs, we have targeted two different breast cancer genes. HER2 and ERα genes were selected as targets to demonstrate the proof of concept. Then, antisense morpholino sequences that were designed for silencing of these genes were hybridized to DNA-tile. HER2 and ERα genes are linked to breast cancer cell proliferation. The increased expression levels of HER2 oncogene are also thought to be related to several cancer types with poor prognostics.35–37
As it is indicated earlier, HER2, an epidermal growth factor receptor (EGFR) family protein, is linked to breast cancer cell proliferation. A decrease in the level of HER2 protein is expected to cause inhibition of cell proliferation.38 Knockdown of HER2 oncogene leads to cell cycle arrest at G0/G1 stage and a decrease in the expressions of a G2/M progression marker cyclin B1 and cdc2 in HER2 overexpressing breast cancer cell lines.39 The estrogen and estrogen receptor have a role in breast cancer formation, and silencing of ERα also induces the decrease in EGFR family gene expression.40 Therefore, HER2 overexpressing BT-474 and ERα overexpressing MCF7 breast cancer cell lines were used in this study to demonstrate the efficiency of DNA-tile-AuNPs. The silencing of HER2 and ERα genes resulted in a decrease in cell proliferation and perturbation of the cell cycle in G0/G1 phase.
The silencing efficiency of DNA-tile-AuNPs was compared with a lipid-based commercial delivery system. At the same morpholino concentrations, DNA-tile-AuNPs nanostructures showed about 30% improved silencing efficiency (in both cell lines and both genes) over liposome-based conventional system.
In conclusion, the low cost and simple construction of DNA-tiles allow the integration of any silencing oligonucleotides into its structure. In addition, DNA-tile-AuNPs nanostructures are suitable for all kind of antisense structures without any discrimination; whether anionic or neutral. The proposed approach can be promising for gene regulation-based personalized treatments. Being able to load drug to the DNA-tile also enables more efficient treatment strategies such as silencing and chemotherapy combination, which is currently under investigation in our laboratories.
For references and to read this article in its entirety click here.
Human Gene Therapy, published by Mary Ann Liebert, Inc., is the premier, multidisciplinary journal covering all aspects of gene therapy. The Journal publishes in-depth coverage of DNA, RNA, and cell therapies by delivering the latest breakthroughs in research and technologies.The above article was first published in the December 2019 issue of Human Gene Therapy. The views expressed here are those of the authors and are not necessarily those of Human Gene Therapy, Mary Ann Liebert, Inc., publishers, or their affiliates. No endorsement of any entity or technology is implied.
The post Silencing Breast Cancer Genes Using Morpholino Embedded DNA-Tile-AuNPs Nanostructures appeared first on GEN – Genetic Engineering and Biotechnology News.












{kind=link}